
The astronomical scale of potential chemical space means that researchers are effectively searching for a single grain of sand on an endless beach, making traditional trial-and-error laboratory experiments both physically and economically impossible in the modern era. As pharmaceutical companies transition toward fully integrated computational workflows, the reliance on high-throughput physical screening is being replaced by sophisticated predictive modeling that can evaluate millions of compounds in seconds. This systemic shift is driven by the realization that chemical libraries have grown to such immense sizes that human intuition and manual testing are no longer sufficient to maintain a competitive development pipeline. By leveraging these advanced digital simulations, scientists can now prioritize drug candidates with unprecedented precision, ensuring that only the most promising molecules move toward expensive clinical validation. This data-centric approach fundamentally changes how researchers define success in the discovery phase, focusing on molecular profiles that offer the best balance of safety and effectiveness from the start.
Decoding Molecular Behavior: The Role of Digital Fingerprints
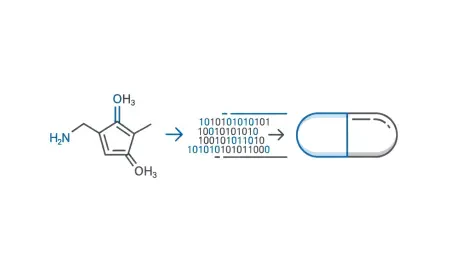
The technical foundation of this prioritization process lies in the ability to translate a molecule’s physical structure into a set of mathematical variables that represent its biological behavior. Using techniques like Quantitative Structure–Activity Relationship (QSAR) models, researchers create complex digital fingerprints known as molecular descriptors. These descriptors account for a wide range of critical traits, such as the arrangement of specific atoms, the molecular weight, and the octanol-water partition coefficient, which indicates how easily a molecule dissolves in lipids versus water. By converting a physical substance into a detailed data set, the modeling software can compare an unknown compound against thousands of previously tested substances to find patterns that suggest high biological activity. This structural analysis allows teams to skip the testing of millions of inactive compounds, focusing instead on structural clusters that show the highest probability of binding effectively with their intended target proteins in the body.
Once these molecular descriptors are established, the next step involves training machine learning algorithms on vast datasets of existing experimental results to ensure the model’s predictive accuracy. These models must be rigorously validated using held-out data to confirm that they can reliably predict how a new, untested compound will interact within a biological system. A crucial aspect of this stage is defining the applicability domain, which ensures the model only makes predictions for chemicals that are sufficiently similar to the training set to be considered reliable. If a compound falls outside this range, the model flags it as an uncertainty, preventing the system from making dangerous guesses that could lead to clinical failure later. This iterative validation ensures that the computational predictions are not just mathematical abstractions but are grounded in the physical reality of biochemical interactions. As training data becomes more diverse from 2026 to 2028, these models are becoming increasingly adept at identifying non-obvious drug candidates.
Optimizing the Virtual Screening: Digital Pipeline Efficiency
The process of virtual screening allows scientists to perform the heavy lifting of drug discovery in silico before a single pipette is touched in a physical laboratory environment. This typically involves a dual-layered approach consisting of ligand-based and structure-based screening to rank potential candidates according to their predicted performance. Ligand-based methods search for structural similarities to molecules already known to be active, whereas structure-based screening uses 3D simulations to see how perfectly a molecule fits into the pocket of a specific protein target. Modern drug discovery workflows frequently utilize a hybrid of these methods to generate a comprehensive ranking system based on binding affinity and selectivity. By simulating these interactions digitally, researchers can effectively filter out molecules that might bind to the wrong targets, which is a major cause of side effects. This selective ranking ensures that the shortlist of candidates provided to lab scientists has already been vetted for basic viability.
Beyond biological activity, modern predictive models have been expanded to include filters for synthetic accessibility to ensure that a theoretically perfect drug can actually be manufactured. It is a common frustration in drug development to find a molecule that performs exceptionally well in a simulation but proves nearly impossible or prohibitively expensive to synthesize in a physical lab. Predictive algorithms now analyze the complexity of the chemical reactions required to build a molecule, flagging those that would require too many steps or rare reagents. This ensures that the prioritization process accounts for the practical realities of pharmaceutical manufacturing and commercial scalability. By integrating these feasibility checks early in the design phase, companies can avoid wasting months of effort on molecules that can never reach the market. This economic filter is just as vital as the biological ones, as it maintains the project’s momentum and ensures the transition from computer to physical manufacturing is as seamless as possible.
Predictive Toxicology: Enhancing Early Safety Assessments
Predictive toxicology has revolutionized the safety assessment phase by allowing researchers to forecast adverse outcomes long before a compound ever enters a clinical trial or even a petri dish. In the past, a significant percentage of drug candidates failed in the late stages of development because of unforeseen liver or heart complications that were not detected during early screening. Today, computational models can identify structural motifs that are known to cause toxicity, such as DNA damage or metabolic interference, allowing for a fail-fast strategy that saves immense resources. By eliminating high-risk candidates at the very beginning of the cycle, the industry can concentrate its investment on molecules that possess a much higher safety profile. This proactive approach to safety screening is not just a cost-saving measure but a fundamental ethical improvement in how medicines are developed. It ensures that the compounds reaching human testing are those with the lowest risk of causing harm, significantly improving the success rates of clinical trials.
The specific technical capabilities of these safety models now include the ability to predict complex interactions like the inhibition of the hERG potassium channel, which is a cause of heart rhythm disruptions. By simulating these specific biological pathways, models can provide detailed insights into how a drug might affect various organ systems without the need for initial animal testing. This level of granularity is achieved by training models on specific toxicological endpoints, allowing them to flag even subtle risks that might have been missed by traditional observational methods. Furthermore, these models are capable of identifying off-target effects where a drug might unintentionally interact with proteins it was not designed to touch. By mapping out these potential interactions in a digital environment, scientists can modify the molecular structure to eliminate the risk while maintaining the drug’s therapeutic benefits. This level of predictive precision is essential for developing next-generation therapies that are as safe as they are effective.
Multi-Parameter Optimization: Balancing Competing Molecular Traits
Selecting a final lead candidate for drug development is rarely a simple task because a molecule that is highly effective at killing a target might also be very difficult for the body to absorb. Multi-parameter optimization (MPO) frameworks address this challenge by allowing researchers to evaluate all critical factors simultaneously rather than analyzing them in isolated silos. By using weighted scoring systems, scientists can determine the optimal trade-off between potency, stability, metabolic clearance, and safety profiles. This holistic view prevents the common pitfall of selecting a compound that excels in one specific laboratory metric but ultimately fails because it cannot reach the intended site of action in the human body. The MPO approach provides a balanced score that reflects the overall likelihood of a compound becoming a successful medicine. This sophisticated ranking system ensures that the development pipeline is populated with robust candidates that have been optimized for the complex environment of the human anatomy.
The most advanced prioritization systems are now moving toward adaptive discovery through the integration of iterative learning algorithms like Bayesian optimization. These systems create a continuous feedback loop where every result from the physical laboratory is immediately fed back into the computer model to refine its subsequent predictions. This means that the modeling process becomes more accurate and intelligent over time as it learns from real-world experimental successes and failures. This constant communication between the computational engine and the laboratory team speeds up the search for high-quality drug leads by reducing the number of redundant or low-value experiments. Rather than following a static plan, the discovery process evolves dynamically based on the data, allowing researchers to pivot quickly when the model identifies a more promising chemical path. This synergy between human expertise and machine intelligence represents the current peak of efficiency in modern pharmaceutical research, ensuring that discovery remains a living process.
Industry Hurdles: Navigating Data Quality and Interpretability
Despite these significant technological leaps, the quality and standardization of available data remain a primary hurdle for the widespread success of predictive modeling in drug discovery. A model is only as reliable as the information used to train it; if the input data is biased, incomplete, or of poor quality, the resulting predictions can be misleading or entirely inaccurate. This often leads to the phenomenon of overfitting, where a model performs perfectly on historical data but fails to predict the behavior of entirely new or diverse chemical structures. Ensuring that data is high-quality and standardized across different laboratories is essential for building models that can be trusted with multimillion-dollar development decisions. Furthermore, the black box nature of some deep learning algorithms can make it difficult for scientists to explain the reasoning behind a specific compound’s ranking. In a regulated industry, transparency is vital for gaining the trust of both internal researchers and external agencies.
To overcome these limitations, the pharmaceutical industry shifted its focus toward developing hybrid models that combined data-driven machine learning with physics-based simulations. These new approaches provided a more complete picture of molecular behavior by grounding statistical predictions in the fundamental laws of chemistry and thermodynamics. Scientific communities also prioritized the standardization of benchmarks and the creation of shared data repositories to improve the overall transparency of computational tools. By adopting more interpretable models, researchers were able to see the specific molecular features that drove a prediction, which allowed for more confident decision-making during the prioritization phase. These advancements ensured that the search for new medicines became more predictable and less reliant on chance, ultimately accelerating the delivery of life-saving therapies to patients. Looking ahead, the focus remained on refining these collaborative frameworks to ensure digital tools continued to provide a reliable bridge to success.